



Whole exome sequencing: principles and diagnostic capabilities

- Authors: Suspitsin E.N1, Tyurin V.I1, Imyanitov E.N1, Sokolenko A.P1
-
Affiliations:
- St Petersburg State Pediatric Medical University, Ministry of Healthcare of the Russian Federation
- Issue: Vol 7, No 4 (2016)
- Pages: 142-146
- Section: Articles
- URL: https://journal-vniispk.ru/pediatr/article/view/5980
- DOI: https://doi.org/10.17816/PED74142-146
- ID: 5980
Cite item
Full Text

Abstract
Keywords
Full Text
Появление высокопроизводительного секвенирования нового поколения (Next Generation Sequencing, NGS) привело к революционному расширению возможностей ДНК-диагностики. Если ранее генетическое тестирование было, как правило, ориентировано на единичные гены, то в настоящее время стала реальной единовременная оценка состояния всего генома.
Термин NGS объединяет различные подходы к крупномасштабной расшифровке генетической информации. Помимо собственно технологии секвенирования, методы NGS отличаются по объему выполняемого анализа. Так, полногеномное секвенирование подразумевает прочтение практически всей геномной ДНК, включая некодирующие последовательности. Таргетное секвенирование, напротив, позволяет сконцентрировать усилия на анализе определенного набора диагностически значимых генов. Наконец, полноэкзомное секвенирование (ПЭС) основано на оценке всех кодирующих последовательностей генома. Именно последний подход, по-видимому, обладает наибольшими перспективами.
Несмотря на то, что суммарная протяженность экзонов, то есть участков ДНК, кодирующих белки, составляет всего 1 % генома, известно, что подавляющее большинство мутаций, имеющих медицинскую значимость, локализуется именно в экзонах [11]. Исходя из этого, для решения диагностических задач более дешевое экзомное секвенирование имеет преимущества перед геномным. С экономической точки зрения альтернативой может служить создание диагностических панелей (мультигенное таргетное секвенирование). Однако такой подход подразумевает, что все гены, мутации которых связаны с риском развития интересующего нас заболевания, уже известны. Полноэкзомное секвенирование лишено данного ограничения, поскольку оно не только позволяет выявлять дефекты известных генов, но и предоставляет шансы открытия новых генетических элементов, вызывающих болезни. Совмещая в себе диагностические и исследовательские возможности, эта технология становится важнейшим инструментом изучения наследственной патологии.
Принципы полноэкзомного секвенирования
Следует заметить, что процедура высокопроизводительного секвенирования параллельно разрабатывалась различными компаниями (Illumina, Roche, Life Technologies), каждая из которых предлагает собственные технологические решения. В любом случае методика подразумевает одновременное секвенирование миллионов коротких фрагментов ДНК с последующей «сборкой» индивидуальных прочтений в геном или экзом.
Главной чертой, отличающей экзомное секвенирование от других видов NGS, является предварительное обогащение ДНК в отношении кодирующих последовательностей. Таким образом, методика ПЭС включает несколько общих этапов: подготовку библиотеки, обогащение, секвенирование и биоинформатическую обработку данных (рис. 1).
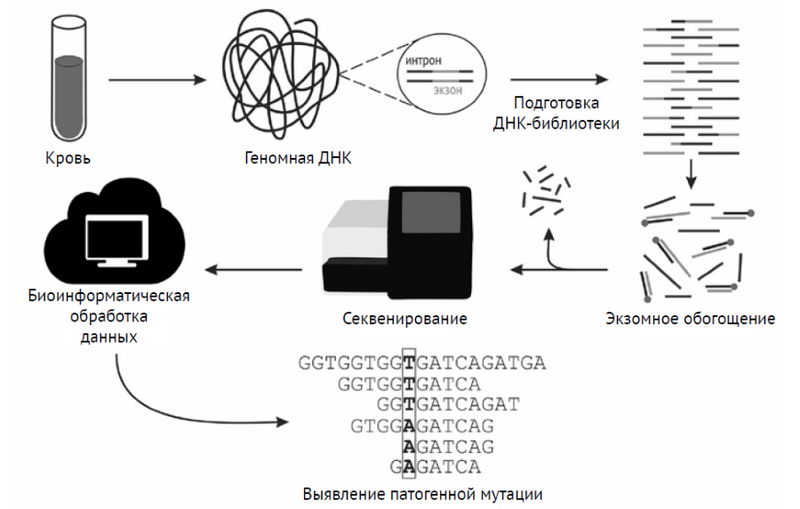
Рис. 1. Основные этапы полноэкзомного секвенирования
В настоящее время наиболее популярным является полноэкзомное секвенирование на платформе фирмы Illumina. Согласно ее протоколу ДНК фрагментируется на короткие кусочки (300–600 пар оснований), к которым пришиваются адаптеры — нуклеотидные последовательности, необходимые для заякоривания на микрочипе и начала процесса секвенирования. Этот этап называется подготовкой библиотеки.
Обогащение по кодирующей последовательности достигается посредством гибридизации ДНК с биотинилированными зондами, специфичными к экзонам. Фрагменты ДНК, связавшиеся с зондами, осаждаются на магнитных частицах, покрытых стрептавидином, и затем используются для секвенирования, а прочие (несвязавшиеся) участки удаляются.
Следующие этапы анализа осуществляются непосредственно в приборе (секвенаторе), в который помещается стеклянный микрочип. На его поверхности фиксированы олигонуклеотиды, последовательность которых комплементарна адаптерам, находящимся на концах фрагментов ДНК. Связывание олигонуклеотидов с адаптерами инициирует проведение так называемой «мостиковой» ПЦР, в результате которой генерируется огромное количество кластеров, или ДНК-клонов.
Размноженные таким образом фрагменты ДНК далее подвергаются секвенированию посредством синтеза. Оно начинается с добавления праймера, комплементарного адаптеру на одном из концов фрагмента; отжиг праймера позволяет ДНК-полимеразе проводить присоединение нуклеотидов. Встраивание нуклеотида, комплементарного матрице, вызывает изменение уровня флуоресцентного сигнала. Последовательное считывание флуоресценции от каждого встроившегося в цепь нуклеотида дает возможность определить состав короткого фрагмента ДНК. В итоге прибор переводит флуоресцентный сигнал на язык букв (нуклеотидов), продуцируя большое число индивидуальных прочтений, которые затем сопоставляются с референсным геномом человека. Выявленные отличия подвергаются аннотации — оценке их влияния на аминокислотную последовательность.
В среднем, в результате секвенирования экзома обнаруживается около 20 000 генетических вариаций [6]; далее они подвергаются фильтрации, задачей которой является отсеивание нейтральных вариантов, не обладающих функциональной значимостью. Этот этап представляет наибольшие сложности, поскольку эффект многих генетических вариантов в отношении функции белка неясен. Для успешного выявления искомых патогенных мутаций необходимо тесное взаимодействие врача-генетика и специалистов в области биоинформатической обработки данных.
Применение в медицине
За относительно недолгое время своего существования ПЭС убедительно продемонстрировало свою эффективность в установлении причин генетических заболеваний [6, 10, 11, 16]. В первую очередь речь идет о болезнях, наследование которых подчиняется законам Менделя. В таких случаях наиболее понятна логика поиска патогенных мутаций: выявленное повреждение должно присутствовать у больных и отсутствовать у здоровых членов семьи. Всего за несколько лет практического применения (2009–2014) экзомное секвенирование позволило обнаружить более 150 новых генов, причастных к развитию редких генетических синдромов [13], и список открытий продолжает постоянно пополняться. Технологический прорыв привел к существенному обогащению представлений о некоторых заболеваниях, этиология которых ранее была неясна: в частности, удалось выявить редкие мутации, приводящие к возникновению шизофрении и расстройствам аутистического спектра [2, 15].
ПЭС с успехом применяется для диагностики наследственных опухолевых синдромов [17], эндокринопатий [4], неврологических заболеваний [7, 9] и первичных иммунодефицитов [3, 12]. Обсуждается также целесообразность использования ПЭС для неонатального скрининга [5].
Применение полноэкзомного секвенирования наиболее оправдано в двух ситуациях — при диагностике редких (орфанных) заболеваний, а также при ДНК-тестировании в случае болезней, обладающих высокой генетической гетерогенностью.
Число нозологических форм орфанных болезней достигает 6000–7000, и большинство из них имеет генетическую природу. Есть данные, что около половины пациентов, несмотря на огромный объем проведенных исследований, так никогда и не получает конкретного диагноза [14]. Этот факт связывают с высокой гетерогенностью и атипичной симптоматикой более или менее известных заболеваний, а также недостаточным знакомством врачей со многими видами редкой патологии. Экзомное секвенирование дает возможность поиска «без гипотезы», то есть без предварительного представления о том, какие именно гены, сигнальные каскады или метаболические пути повреждены в организме пациента. В этом случае оно служит исследовательским инструментом, результатом применения которого может стать открытие нового гена или не описанного ранее синдрома. Как показывает опыт, завершение «диагностической одиссеи», то есть обретение пациентом конкретного диагноза, всегда приносит семье существенное облегчение.
Учитывая исключительную редкость некоторых заболеваний, успех поиска зависит от рациональной организации исследования. Выявить патогенную мутацию относительно просто, если удается найти семью, у нескольких членов которой присутствуют сходные клинические признаки. Если это невозможно, то можно попытаться набрать в исследование спорадические случаи заболевания с похожей симптоматикой.
Нередко заболевания генетической природы возникают вследствие так называемых мутаций de novo — наследственных дефектов, которые отсутствуют в экзоме родителей и возникают непосредственно у ребенка вследствие естественного мутагенеза. Для того чтобы выявить такую мутацию, требуется провести экзомное секвенирование триады «здоровый отец — здоровая мать — больной ребенок».
В процессе диагностики болезней с высокой генетической гетерогенностью использование ПЭС может быть продиктовано соображениями экономической целесообразности. Например, известно около 250 генов, причастных к развитию несиндромальной тугоухости; помимо этого, нарушения слуха входят в структуру примерно 400 различных синдромов [13]. В подобной ситуации выборочный анализ отдельных генов крайне редко приводит к успеху.
Преимущества полноэкзомного секвенирования продемонстрированы авторами при диагностике синдрома Барде – Бидля — аутосомно-рецессивного наследственного заболевания, основными клиническими проявлениями которого являются ожирение, гипогонадизм, пигментный ретинит и патология почек. Сложность генетического тестирования в данном случае связана с тем, что на сегодняшний момент известно не менее 19 генов, ответственных за развитие данной болезни [1]; при этом каузативные генетические повреждения удается выявить лишь у 80 % больных, что свидетельствует о существовании других, еще не открытых генов. В ходе наших исследований секвенирование семейных случаев синдрома Барде – Бидля выявило у больных не описанную ранее мутацию гена BBS7 в гомозиготном состоянии. Поскольку родители больных детей не являлись родственниками, возникло предположение, что найденная мутация может представлять вариант, встречающийся в российской популяции с существенной частотой. Для проверки этой гипотезы были протестированы 2823 здоровых донора; носительство мутации было выявлено у двух индивидуумов (0,07 %). Таким образом, выявленный аллель безусловно заслуживает внимания при ДНК-диагностике российских пациентов с подозрением на синдром Барде – Бидля [18].
Несмотря на то, что современные методы ПЭС позволяют установить точный диагноз лишь у 16–50 % пациентов с генетическими заболеваниями [14, 19], это грандиозный шаг вперед по сравнению с эффективностью традиционных подходов (кариотипирование, секвенирование по Сэнгеру и т. п.). Несомненно, процент успешной диагностики будет расти по мере совершенствования технологии экзомного обогащения, секвенирования и алгоритмов обработки данных.
В настоящее время широкому использованию ПЭС в лабораторной диагностике препятствует ряд факторов: высокая стоимость реагентов и оборудования, сложность и длительность биоинформатической обработки данных, а также необходимость подтверждения обнаруженных мутаций другими методами.
Развитие технологий высокопроизводительного секвенирования неизбежно приведет к изменениям в работе врачей-генетиков. Сейчас от них требуется в первую очередь умение поставить диагноз редкого генетического синдрома, ориентируясь на особенности фенотипа. Учитывая огромное количество существующих нозологических форм, накопление достаточного опыта обязательно требует многолетней практической работы. По-видимому, ценность непосредственно клинических навыков будет в среднесрочной перспективе несколько снижаться, тогда как на первый план выйдут навыки, связанные с правильной оценкой функциональной значимости выявленных мутаций.
Нельзя не упомянуть и этические проблемы, связанные с применением ПЭС. Дело в том, что помимо выявления непосредственно интересующего нас генетического дефекта, секвенирование экзома может в качестве случайных находок обнаружить и другие значимые с медицинской точки зрения мутации. Возникает вопрос, нужно ли доводить эту информацию до сведения пациента и его родственников, и если да, то в какой форме. Специалистами Американского колледжа медицинских генетиков (ACMG) разработан список из 56 генов, об обнаружении мутаций в которых рекомендуется непременно сообщить пациентам, поскольку в таких случаях ранняя, досимптоматическая диагностика принесет пользу [8]. Однако на сегодняшний день единая точка зрения на эту проблему пока не выработана.
Работа поддержана грантом РНФ 14-25-00111.
About the authors
Evgeny N Suspitsin
St Petersburg State Pediatric Medical University, Ministry of Healthcare of the Russian Federation
Author for correspondence.
Email: evgeny.suspitsin@gmail.com
MD, PhD, Associate Professor, Department of Medical Genetics Russian Federation
Vladislav I Tyurin
St Petersburg State Pediatric Medical University, Ministry of Healthcare of the Russian Federation
Email: tyurinvladislav@gmail.com
Resident doctor, Department of Medical Genetics Russian Federation
Evgeny N Imyanitov
St Petersburg State Pediatric Medical University, Ministry of Healthcare of the Russian Federation
Email: evgeny@imyanitov.spb.ru
MD, PhD, Dr Med Sci, Professor, Head, Department of Medical Genetics Russian Federation
Anna P Sokolenko
St Petersburg State Pediatric Medical University, Ministry of Healthcare of the Russian Federation
Email: annasokolenko@mail.ru
MD, PhD, Associate Professor, Department of Medical Genetics Russian Federation
References
- Aldahmesh MA, Li Y, Alhashem A, et al. IFT27, encoding a small GTPase component of IFT particles, is mutated in a consanguineous family with Bardet-Biedl syndrome. Hum Mol Genet. 2014;23:3307-3315. doi: 10.1093/hmg/ddu044.
- Baker E, Jeste SS. Diagnosis and management of autism spectrum disorder in the era of genomics: rare disorders can pave the way for targeted treatments. Pediatr Clin North Am. 2015;62(3):607-618. doi: 10.1016/j.pcl.2015.03.003.
- Chinen J, Notarangelo LD, Shearer WT. Advances in basic and clinical immunology in 2014. J Allergy Clin Immunol. 2015;135(5):1132-41. doi: 10.1016/j.jaci.2015.02.037.
- de Bruin C, Dauber A. Insights from exome sequen¬cing for endocrine disorders. Nat Rev Endocrinol. 2015;11(8):455-64. doi: 10.1038/nrendo.2015.72.
- Francescatto L, Katsanis N. Newborn screening and the era of medical genomics. Semin Perinatol. 2015;39(8):617-22. doi: 10.1053/j.semperi.2015.
- 010.
- Gilissen C, Hoischen A, Brunner HG, Veltman JA. Unlocking Mendelian disease using exome sequencing. Genome Biol. 2011;12:228. doi: 10.1186/gb-2011-12-9-228.
- Ghaoui R, Cooper ST, Lek M, et al. Use of Whole-Exome Sequencing for Diagnosis of Limb-Girdle Muscular Dystrophy: Outcomes and Lessons Learned. JAMA Neurol. 2015;5:1-9. doi: 10.1001/jamaneurol.2015.2274.
- Green RC, Berg JS, Grody WW, et al. ACMG recommendations for reporting of incidental findings in clinical exome and genome sequencing. Genet Med. 2013;15(7):565-74. doi: 10.1038/gim.2013.73.
- Hoischen A, Krumm N, Eichler EE. Prioritization of neurodevelopmental disease genes by discovery of new mutations. Nat Neurosci. 2014;17(6):764-72. doi: 10.1038/nn.3703.
- Ku CS, Cooper DN, Polychronakos C, et al. Exome sequencing: dual role as a discovery and diagnostic tool. Ann Neurol. 2012;71:5-14. doi: 10.1002/ana.22647.
- Ng SB, Buckingham KJ, Lee C, et al. Exome sequencing identifies the cause of a mendelian disorder. Nat Genet. 2010;42:30-35. doi: 10.1038/ng.499.
- Platt C, Geha RS, Chou J. Gene hunting in the genomic era: approaches to diagnostic dilemmas in patients with primary immunodeficiencies. J Allergy Clin Immunol. 2014;134:262-8. doi: 10.1016/j.jaci.2013.08.021.
- Rabbani B, Tekin M, Mahdieh N. The promise of whole-exome sequencing in medical genetics. J Hum Genet. 2014;59:5-15. doi: 10.1038/jhg.2013.114.
- Shashi V, McConkie-Rosell A, Rosell B, et al. The utility of the traditional medical genetics diagnostic evaluation in the context of next-generation sequencing for undiagnosed genetic disorders. Genet Med. 2014;16(2):176-82. doi: 10.1038/gim.2013.99.
- Schreiber M, Dorschner M, Tsuang D. Next-generation sequencing in schizophrenia and other neuropsychiatric disorders. Am J Med Genet. Part B: Neuropsychiatr Genet. 2013;162(7):671-8. doi: 10.1002/ajmg.b.32156.
- Singleton AB. Exome sequencing: a transformative technology. Lancet Neurol. 2011;10:942-946. doi: 10.1016/S1474-4422(11)70196-X.
- Sokolenko AP, Suspitsin EN, Kuligina ESh, et al. Identification of novel hereditary cancer genes by whole exome sequencing. Cancer Lett. 2015;369(2):274-88. doi: 10.1016/j.canlet.2015.09.014.
- Suspitsin EN, Sokolenko AP, Lyazina LV, et al. Exome Sequencing of a Family with Bardet-Biedl Syndrome Identifies the Common Russian Mutation c.1967_1968delTAinsC in BBS7. Mol Syndromol. 2015;6(2):96-8. doi: 10.1159/000371408.
- Valencia CA, Husami A, Holle J, et al. Clinical Impact and Cost-Effectiveness of Whole Exome Sequencing as a Diagnostic Tool: A Pediatric Center’s Experience. Front Pediatr. 2015;(3):67. doi: 10.3389/fped.2015.00067.
Supplementary files

