



")

Влияние внутриклеточных доменов химерных антигенных рецепторов на свойства CAR-T-клеток
- Авторы: Волков Д.В.1, Степанова В.М.1, Ярошевич И.А.1,2, Габибов А.Г.1, Рубцов Ю.П.1
-
Учреждения:
- ГНЦ Институт биоорганической химии им. академиков М.М. Шемякина и Ю.А. Овчинникова РАН
- Московский государственный университет имени М.В. Ломоносова
- Выпуск: Том 17, № 3 (2025)
- Страницы: 4-17
- Раздел: Обзоры
- URL: https://journal-vniispk.ru/2075-8251/article/view/348459
- DOI: https://doi.org/10.32607/actanaturae.27728
- ID: 348459
Цитировать

Аннотация
Технология модификации Т-клеток химерными антигенными рецепторами (CAR – chimeric antigen receptor) расширила возможности терапии онкогематологических заболеваний и изменила вектор развития исследований в области инженерии иммунных клеток и иммунотерапии. К сожалению, успехи терапии Т-клетками, модифицированными химерными антигенными рецепторами (CAR-T-cell – chimeric antigen receptor modified T cell), в отдельных случаях онкогематологических заболеваний и солидных опухолей ограничены рядом факторов, а именно: (1) избыточным или недостаточным ответом CAR-T-клеток, развивающимся как из-за резистентности опухолевых клеток или их микроокружения, так и за счет неоптимальной структурно-функциональной организации химерного рецептора; (2) не самым функциональным фенотипом готового CAR-T-клеточного продукта, формирование которого является прямым следствием процесса получения CAR-T-клеток и их экспансии; (3) отсутствием адекватной системы управления CAR-T-клетками после введения пациенту. Поэтому актуальные задачи современных исследований включают оптимизацию структуры CAR и технологий их получения, а также дополнительные модификации CAR-T-клеток. Одно из главных направлений повышения эффективности терапии с помощью CAR-T-клеток – это оптимизация структуры CAR с целью улучшения функционирования модифицированных клеток. С момента появления первых CAR-T-клеток создано пять поколений CAR, в которых использованы как новые сочетания сигнальных и структурных доменов в одной молекуле, так и новые системы из нескольких химерных молекул, представленных одновременно на поверхности Т-клеток. Рациональная комбинация составных частей CAR должна обеспечивать высокую чувствительность рецептора к антигену, образование устойчивого иммунного синапса (ИС), эффективную костимуляцию и продуктивную активацию CAR-T-клетки. Сочетание современных технологий – машинного обучения для предсказания трехмерной структуры и свойств биополимеров, а также высокопроизводительного секвенирования и омиксных технологий – открывает новые горизонты для направленной модификации структуры CAR. Ключевым становится выбор конкретных модификаций и сочетаний костимулирующих и сигнальных доменов с целью повышения цитотоксичности, пролиферации и персистенции CAR-T-клеток. В представленном обзоре обсуждаются последние достижения в области оптимизаций CAR с акцентом на изменения, которые должны улучшать функции терапевтических CAR-T-клеток.
Ключевые слова
Полный текст
СПИСОК СОКРАЩЕНИЙ
CAR – химерный антигенный рецептор; CAR-T-клетки – Т-клетки, модифицированные химерным антигенным рецептором; ИС – иммунный синапс; ОАА – опухоль-ассоциированный антиген; FAS-L – Fas-лиганд; FAS – Fas-рецептор; ICD – внутриклеточный домен; scFv – одноцепочечный вариабельный фрагмент; VHH – вариабельный фрагмент наноантитела; CD – кластер дифференцировки; BCMA – антиген созревания B-клеток; IgSF – суперсемейство иммуноглобулинов; TNFRSF – суперсемейство рецепторов фактора некроза опухоли; TNFSF – суперсемейство фактора некроза опухоли; АПК – антигенпрезентирующая клетка; ГКГСI/II – главный комплекс гистосовместимости I и II типа; ТКР – Т-клеточный рецептор; α, β – распознающие цепи Т-клеточного рецептора; ζ, γ, δ, ε – белки группы CD3 Т-клеточного рецептора; ITAM – иммунорецепторный тирозиновый активирующий мотив; АК – аминокислота; Y – тирозин; Tn-клетки – наивные Т-клетки; ICOSL – лиганд рецептора ICOS; ИЛ – интерлейкин; ИФН-γ – интерферон-гамма; TNF-α – фактор некроза опухоли-альфа; Treg-клетки – регуляторные Т-клетки; FAP – белок, активирующий фибробласты; Th-клетки – Т-хелперы; Tm-клетки – Т-клетки памяти; TRAF – рецепторы, связывающие факторы, ассоциированные с рецептором фактора некроза опухоли; Tcm-клетки – Т-клетки центральной памяти; HVEM – медиатор проникновения вируса герпеса; Tem-клетки – Т-клетки эффекторной памяти; GITR – глюкокортикоид-индуцированный TNF-рецептор-связанный белок; BRS – участки некоторых молекул CD3, обогащенные положительно заряженными АК; RK – рецепторная киназа; PRS – богатая пролином последовательность CD3ε; PKC – протеинкиназа C; bCAR – химерные антигенные рецепторы, в которых сигнальные домены представлены частями различных внутриклеточных сигнальных партнеров ТКР; ZAP70KD – киназный домен ZAP70.
ВВЕДЕНИЕ
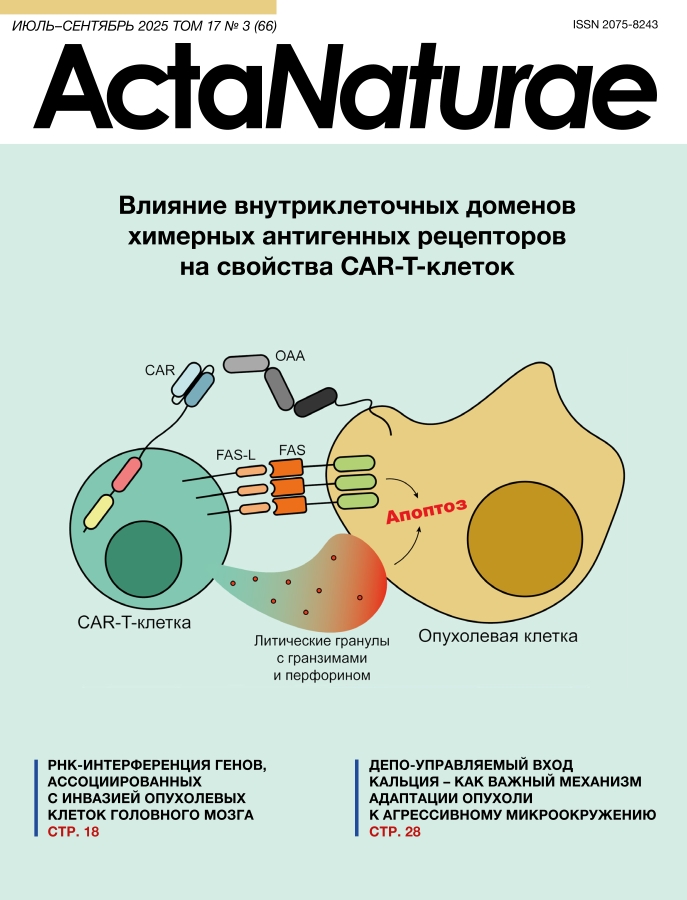
Традиционные методы лечения опухолевых заболеваний – химио- и лучевая терапия – в настоящее время достаточно часто сочетаются с относительно новыми – иммунотерапевтическими. К ним относятся терапия препаратами моноклональных антител, биспецифическими активаторами Т-клеток, а также клеточная терапия, в том числе CAR-T-клетками, на которых сфокусирован наш обзор. Постепенный сдвиг в сторону более специфичных или, как их называют, таргетных методов лечения объясняется низкой эффективностью и тяжелыми побочными реакциями традиционных методов терапии (например, системной генотоксичностью) [1], а также возросшим потенциалом новых методов терапии, что особенно ярко демонстрирует технология CAR-T-терапии в случае онкогематологических заболеваний [2]. Принцип действия CAR-T-терапии основан на распознавании поверхностных маркеров опухолевых клеток, что позволяет специфично воздействовать на них цитотоксическими CAR-Т-клетками (рис. 1).

Рис. 1. Механистический принцип CAR-T-терапии. Взаимодействие CAR-T-клетки с опухолевой клеткой обеспечивается специфичным распознаванием опухоль-ассоциированного антигена (ОАА) химерным рецептором. Это приводит к активации цитотоксических функций CAR-T-клетки, опосредованных выделением литических гранул с гранзимами и перфорином, а также взаимодействием Fas-лиганда (FAS-L) и Fas-рецептора (FAS). В результате наблюдается апоптоз опухолевой клетки
Это воздействие обеспечивается CAR, который состоит из трех основных доменов – внеклеточного, ответственного за распознавание антигена и подвижность распознающей части, трансмембранного, участвующего в формировании ИС, и внутриклеточного, содержащего костимулирующие и сигнальный домены, которые определяют весь спектр ответов CAR-T-клеток на специфическую активацию при связывании антигена. При этом внеопухолевая токсичность таких иммунотерапевтических препаратов существенно ниже по сравнению с традиционной терапией [3]. Кроме того, CAR-T-терапия, в случае успеха, позволяет сформировать популяцию специфичных клеток памяти, что обеспечивает долгую ремиссию [4]. К сожалению, несмотря на отдельные успехи в применении CAR-T-клеток, остаются пациенты, которым CAR-T-терапия в существующем виде помогает лишь временно, в силу недостаточной персистенции или цитотоксичности высокоперсонализированных клеточных препаратов. Поэтому усилия исследователей направлены на повышение эффективности CAR-T-терапии. Одним из ключевых факторов этого является эффективность передачи сигнала от CAR на мембране внутрь клеток, что вызывает активацию транскрипционных программ, ответственных за цитотоксичность, выживание активированных клеток, их пролиферацию, секрецию цитокинов и литических гранул, метаболизм и другие функции. За передачу сигнала отвечают внутриклеточные домены (ICD – intracellular domain) CAR, оптимизации структуры которых посвящен данный обзор.
КОСТИМУЛИРУЮЩИЕ ДОМЕНЫ
В CAR-T-клеточных препаратах, одобренных для клинического применения, роль костимулирующих доменов выполняют внутриклеточные части достаточно хорошо изученных костимулирующих мембранных молекул Т-клеток – CD28 и 4-1BB [5] (рис. 2).

Рис. 2. Одобренные к применению CAR-T-клеточные препараты. Показаны основные домены, обеспечивающие распознавание и передачу сигнала внутриклеточным партнерам. scFv – одноцепочечный вариабельный фрагмент; VHH – вариабельный фрагмент наноантитела; CD – кластер дифференцировки; BCMA – антиген созревания B-клеток
Эти мембранные белки относятся к двум суперсемействам – иммуноглобулиновому (IgSF – immunoglobulin superfamily) и семейству рецепторов фактора некроза опухоли (TNFRSF – tumor necrosis factor receptor superfamily) соответственно. На разных стадиях разработки находятся CAR с другими костимулирующими доменами, которые также входят в эти семейства – ICOS, OX40, CD27 и др. (рис. 3).

Рис. 3. Основные суперсемейства Т-клеточных костимулирующих рецепторов. Показана общая схема активации Т-клеток, для которой необходим основной сигнал и костимулирующие, обеспечиваемые взаимодействием как ТКР с главным комплексом гистосовместимости, так и активирующих рецепторов – представителей IgSF и TNFRSF – со своими лигандами. У рецепторов обозначены аминокислотные последовательности основных сигнальных мотивов. АПК – антигенпрезентирующая клетка; ГКГСI/II – главный комплекс гистосовместимости I и II типа; IgSF – суперсемейство иммуноглобулинов; TNFRSF – суперсемейство рецепторов фактора некроза опухоли; TNFSF – суперсемейство фактора некроза опухоли; ТКР – Т-клеточный рецептор; α, β – распознающие цепи Т-клеточного рецептора; ζ, γ, δ, ε – белки группы CD3 Т-клеточного рецептора; ITAM – иммунорецепторный тирозиновый активирующий мотив
Суперсемейство иммуноглобулинов
Среди рецепторов, относящихся к IgSF, молекулы CD28 и ICOS функционируют как стимуляторы Т-клеток за счет консервативного мотива YXXM (X – любая аминокислота (АК)), содержащего остаток тирозина (Y), который при активации фосфорилируется, что стимулирует взаимодействие с внутриклеточными сигнальными партнерами, в том числе с различными киназами.
CD28. CD28 был первым костимулятором, использованным для получения модифицированных T-клеток, содержащих CAR второго поколения [6], которые обладали преимуществами в персистенции и секреции цитокинов по сравнению с клетками первого поколения, содержащими только сигнальный домен CD3ζ [7]. Сигналы CD28 критически важны для активации наивных Т-клеток (Tn – naive T cell), так как предохраняют эти клетки от анергии [8], а также поддерживают такие процессы, как секреция цитокинов, пролиферация Т-клеток и их дифференцировка в эффекторные клетки. Взаимодействие CD28 с несколькими лигандами, в числе которых CD80 (B7-1), CD86 (B7-2) и B7-H2 (ICOSL), который одновременно является лигандом ICOS, приводит к его активации. Функциональные мотивы во внутриклеточной части CD28 – проксимальные (YMNM, PRRP) и дистальный (PYAP) (рис. 3) – связывают киназы с доменами SH2 и/или SH3 (YMNM – SHIP1, SLP76, GRAP, CBL, PI3K, GRB2 и GADS; PRRP – ITK; PYAP – PDK1, PKCθ, GRB2, STS1/2, CIN85, CD2AP, LCK и FLNA). Фиксация киназ на костимуляторе обеспечивает изменение их конформации, активацию и последующие взаимодействия киназ с нижележащими участниками сигнальных каскадов, которые, с одной стороны, приводят к активации транскрипционных факторов NFAT, AP-1 и NF-ϰB, ассоциированных с синтезом интерлейкина-2 (ИЛ-2) и стимуляцией Bcl-XL, а с другой, стимулируют метаболизм Т-клеток, усиливая аэробный гликолиз, поступление питательных веществ и анаболические процессы [8–10].
В конструкции CAR используют ICD CD28, который при связывании антигена с химерным рецептором ведет к активации пути PI3K/AKT, усиливая аэробный гликолиз, который позитивно влияет на эффекторные свойства Т-клеток [11]. В то же время высокий уровень гликолиза провоцирует истощение клеток и снижает их персистенцию [12]. Для преодоления этого пробуют вносить мутации в функциональные мотивы CD28. При этом мутации в каждом мотиве могут модифицировать характеристики полученных CAR-T-клеток. Так, в модели ксенотрансплантата панкреатической опухоли замена мотива YMNM на YMFM в SS1-CAR-T-клетках на основе CD28, нацеленных на мезотелин, снижает взаимодействие CD28 с GRB2, что уменьшает передачу сигналов через VAV1, снижает кальциевый ток и гиперактивацию NFAT, уменьшая истощение и дисфункцию Т-клеток и повышая их персистенцию и противоопухолевую эффективность [12]. CD28 с заменой мотивов PRRP и YMNM на ARRA и YFNM соответственно, усиливает секрецию клетками интерферона-гамма (ИФН-γ) и фактора некроза опухоли-альфа (TNF-α – tumor necrosis factor alpha), снижает уровень связанного с истощением транскрипционного фактора Nur77 и повышает цитотоксичность CD19-CAR-T-клеток, что, в результате, обеспечивает устойчивое ингибирование развития опухоли у мышей [13]. Kofler и соавт. показали, что замена в CD28 участка PYAPP на AYAAA нарушает взаимодействие мотива PYAP с киназой LCK, что снижает секрецию ИЛ-2 и подавляет зависимую от него сигнализацию, а также ослабляет ИЛ-2-зависимую пролиферацию внутриопухолевых регуляторных Т-клеток (Treg – regulatory T cells), усиливая тем самым противоопухолевую активность таких CAR-T-клеток в отношении солидных опухолей с высокой инфильтрацией Treg-клетками [14]. Кроме того, эта модификация CD28 усиливает пролиферацию, метаболизм, активацию и цитотоксичность CAR-T-клеток, нацеленных на белок, активирующий фибробласты (FAP – fibroblast activation protein). Такие клетки эффективно элиминируют опухоли в комбинации с ингибиторами иммунных контрольных точек и длительно персистируют в гуманизированных мышах с ксенотрансплантатом и у пациентов со злокачественной плевральной мезотелиомой, демонстрируя высокий уровень безопасности [15]. Так как ICD CD28 часто включают в состав CAR вместе с трансмембранным доменом, стоит отметить, что за счет этого CAR может образовывать гетеродимеры с эндогенным CD28 [16], что приводит как к тоническому сигналингу, так и к усилению эффекторных функций соответствующих CAR-T-клеток.
ICOS. Этот рецептор слабо экспрессируется в Tn-клетках до активации Т-клеточного рецептора (ТКР), однако после активации экспрессия усиливается в течение нескольких часов [17]. ICOS активирует взаимодействие с лигандом – ICOSL, которое поддерживает жизнедеятельность Т-клеток, стимулируя пролиферацию и дифференцировку по сходным с CD28 механизмам. Эти рецепторы различаются по влиянию на синтез и секрецию цитокинов. Так, CD28 стимулирует продукцию ИЛ-2, а ICOS – ИЛ-10, характерного в большей степени для Treg-клеток [18]. Кроме ИЛ-10, ICOS также стимулирует секрецию ИФН-γ, TNF-α, ИЛ-5, ИЛ-13 и ИЛ-17, что усиливает эффекторные свойства Т-клеток и дифференцировку наивных Т-хелперов (Th – T helper cell) в эффекторные клетки подтипов Th1, Th2 и Th17 [18, 19]. Как и CD28, ICOS активирует экспрессию Bcl-XL, что повышает жизнеспособность Т-клеток [20]. Функциональный тирозинсодержащий мотив ICOS YMFM (рис. 3) взаимодействует с регуляторной субъединицей PI3K p50α, что приводит к более сильной активации PI3K по сравнению с эффектом CD28 [21]. В результате стимуляция ICOS приводит к фосфорилированию киназ AKT, PDK1, ERK1/2, p38 MAPK и активации транскрипционных факторов NFAT и NF-ϰB, что отличает ICOS от CD28, который также вовлекает в сигнальный каскад киназу JNK и активирует транскрипционный фактор c-Jun [17].
Функциональную активность CAR-T-клеток на основе ICOS первыми показали Shen и соавт., которые применили CAR-T-клетки в лечении глиобластомы в мышиной модели [22]. Предполагается, что именно поляризация CD4+ CAR-Т-клеток в сторону Th1 и Th17 за счет вовлечения PI3K/AKT, p38 MAPK (и других механизмов) усиливает их персистенцию [23], что повышает противоопухолевую активность и CD8+ CAR-T-клеток [23]. Перед введением животным CD4+ mesoCAR-T-клеток, нацеленных на мезотелин, Wyatt и соавт. проводили низкоинтенсивную стимуляцию этих клеток с помощью магнитных шариков, покрытых антителами к CD3 и ICOS (соотношение шарики : клетки = 1 : 10), параллельно добиваясь поляризации клеток в Th17 с помощью коктейля цитокинов. Такая обработка обеспечивала (по сравнению со стимуляцией CD3 и CD28) получение менее дифференцированных CD4+ CAR-T-клеток и сдвиг их метаболизма в сторону окислительного фосфорилирования, что характерно для Т-клеток памяти (Tm – memory T cell). Это подчеркивает преимущество именно ICOS-направленной стимуляции CD4+ mesoCAR-Т-клеток. Такая подготовка приводила к более эффективной элиминации опухоли у мышей с мезотелиомой с помощью комбинации CD4+ Th17 mesoCAR-T-клеток и CD8+ mesoCAR-T-клеток [24] по сравнению со стандартно активированными CAR-T-клетками. Эти данные подчеркивают неодинаковую роль разных костимуляторов в зависимости от типа CAR-Т-клеток – CD4+ или CD8+. Оптимальная костимуляция может быть обеспечена с помощью модификации CD4+ и CD8+ Т-клеток генами CAR с разными костимулирующими доменами, что необходимо учитывать для получения наиболее эффективного CAR-T-клеточного продукта. Исследование эффекта костимуляции CAR-T-клеток с помощью ICOS показало, что замена YMFM на FMFM приводит к нарушению костимуляции CAR-T-клеток через ICOS. Это ведет к низкой секреции указанных ранее цитокинов CAR-T-клетками [25]. Модификации ICD ICOS, повышающие его эффективность, пока не описаны.
Суперсемейство рецепторов фактора некроза опухоли
В состав этого обширного суперсемейства входит порядка 30 рецепторов, которые делятся на три группы: рецепторы, связывающие факторы, ассоциированные с рецептором фактора некроза опухоли (TRAF – TNF receptor-associated factor) (1); рецепторы смерти (2) и молекулы с нефункциональным ICD или без него (3) [26]. В структуре CAR к этому моменту использовали внутриклеточные части только рецепторов группы (1) – это 4-1BB, OX40, CD27, HVEM, TNFRSF18. Консервативные мотивы рецепторов этой группы включают TRAF-связывающие (P/A/S/T)X(E/Q)E и PXQXXD (X – любая АК) [27].
4-1BB. 4-1BB очень часто включают в CAR при создании CAR-T-клеточных препаратов. Четыре из шести одобренных CAR-T-клеточных продуктов содержат в составе CAR именно 4-1BB [2]. Это не случайно, так как 4-1BB – это один из ключевых маркеров активации Т-клеток, который, благодаря взаимодействию с лигандом 4-1BBL и привлечению разных TRAF, инициирует сигнальные пути p38 MAPK, AKT и ERK. Это приводит к активации транскрипции с NF-ϰB-зависимых промоторов, что повышает продукцию сурвивина, Bcl-XL, Bfl-1, Bcl-2 и снижает уровень Bim [28, 29]. Кроме того, сигнал от 4-1BB увеличивает количество митохондрий и трансмембранный потенциал, что усиливает аэробные процессы в Т-клетках, повышая их эффекторные функции [30]. TRAF-связывающие мотивы 4-1BB – это QEED и EEEE (рис. 3), с которыми взаимодействуют TRAF1, TRAF2, TRAF3 и TRAF5 [31].
Включение 4-1BB в структуру CAR увеличивает персистенцию CAR-T-клеток, которые фенотипически становятся очень похожими на Т-клетки центральной памяти (Tcm – central memory T cell). Они слабо экспонируют на своей поверхности один из наиболее характерных маркеров истощения Т-клеток – PD-1 [32, 33]. Отчасти это объясняет характер метаболизма клеток, который при костимуляции через 4-1BB смещается в сторону усиления митохондриальных процессов и повышенного синтеза митохондрий. Кроме того, в клетках, CAR которых содержат 4-1BB, экспрессия генов антиапоптотических факторов повышена, а проапоптотических снижена. В то же время CAR-T-клетки слабее активируются при костимуляции 4-1BB, чем CD28 [34]. Это объясняется привлечением тандема фосфатаз THEMIS-SHP1, который формирует комплекс с 4-1BB за счет мотива из 10 АК на C-конце 4-1BB. Образующийся комплекс препятствует фосфорилированию сигнального домена CAR – CD3ζ. Мутации в QEED и EEEE снижают секрецию цитокинов, долю Tcm-клеток и противоопухолевую активность CAR-T-клеток [25, 35]. Однако опубликованы данные, согласно которым включение 4-1BB в состав CAR приводит к повышенной агрегации CAR-T-клеток, что снижает их жизнеспособность [36]. Примечательно, что удаление упомянутых 10 АК с C-конца 4-1BB в этом случае препятствует агрегации и восстанавливает функциональность CAR-T-клеток. Кроме того, установлено, что 4-1BB индуцирует тонический сигналинг, который способствует апоптозу CAR-T-клеток [37]. Снижение продукции таких CAR позволяет нормализовать активность CAR-T-клеток.
OX40. OX40 – это костимулирующий рецептор, который появляется на поверхности Тn-клеток только после их активации. Связывание ОХ40 со своим лигандом OX40L обеспечивает привлечение TRAF2, 3, 5 через мотив PIQEE (рис. 3) [38, 39]. Адаптеры TRAF2, 3, 5 индуцируют сигнальный путь NF-ϰB, который способствует синтезу в клетках антиапоптотических факторов Bcl-XL и Bfl-1 [40]. Активируются также киназы PI3K/AKT, вовлеченные в синтез сурвивина и киназы Аврора B, что ингибирует апоптоз и способствует пролиферации T-клеток [41, 42].
Костимуляция CAR-T-клеток второго поколения через OX40 способствует более длительной их персистенции по сравнению с клетками, в которых за костимуляцию в CAR отвечают CD28 и 4-1BB. При этом противоопухолевая активность CAR-T-клеток in vivo практически не зависит от костимулирующего домена CAR. In vitro CAR-T-клетки, в которых за костимуляцию отвечает OX40, лучше убивают таргетные клетки [43]. Транскриптомный анализ таких CAR-T-клеток выявил в них повышенную экспрессию генов, ответственных за репарацию ДНК, окислительное фосфорилирование, ингибирование апоптоза, дифференцировку в клетки памяти и пролиферацию. Данные о так называемой «специализации» OX40 и 4-1BB показывают, что 4-1BB преимущественно стимулирует формирование CD8+ Тm-клеток, а OX40 – CD4+ Tm-клеток [39]. Вместе с информацией о том, что ICOS способствует дифференцировке CD4+ Т-клеток в эффекторы типа Th1, Th2 и Th17, это, по-видимому, свидетельствует о том, что наиболее рациональная комбинация для костимуляции CD4+ CAR-T-клеток включает ICOS и OX40. В то же время комбинация CD28 и 4-1BB, возможно, будет более подходящим вариантом для костимуляции CD8+ CAR-T-клеток.
CD27. CD27 связывает лиганд CD70 и способствует пролиферации и дифференцировке Т-клеток за счет активации сигнальных путей NF-ϰB, PI3K/AKT, SAPK/JNK [44, 45]. Стимуляция через CD27 приводит к уменьшению количества FasL в CD4+ Т-клетках и повышению Bcl-XL и Pim-1 в CD8+ Т-клетках, что ингибирует апоптоз и способствует гликолизу в CD8+ Т-клетках [46–48]. Тем самым CD27 поддерживает пролиферацию и жизнеспособность эффекторных Т-клеток и формирование пула Tm-клеток на стадии первичной активации Tn-клеток, во время клональной экспансии и на эффекторной стадии (например, в опухоли). Для взаимодействия с TRAF2, 3, 5 CD27 использует функциональный мотив PIQED(YR) и, возможно, EEEG (рис. 3) [45, 49]. Уникальное свойство CD27, отличающее его от представителей семейства TNFRSF, – формирование гомодимеров за счет дисульфидных мостиков [49]. Именно в этой форме CD27 присутствует на поверхности покоящихся Т-клеток, в то время как их продолжительная активация повышает долю мономерной формы, что, вероятно, защищает Т-клетки от включения костимуляторов при спонтанной активации.
Изучение костимулирующего потенциала CD27 показало, что CD27 CAR-T-клетки способны более эффективно уничтожать опухоли, подобно тому, как это делают CAR-T-клетки с костимуляторами CD28 или 4-1BB, чем CAR-T-клетки первого поколения. Продолжительность персистенции CAR-T-клеток c костимулятором CD27 сходна с персистенцией CAR-T-клеток с костимулятором 4-1BB [50, 51]. В то же время прямое сравнение способности CAR-T-клеток второго поколения с 4-1BB или CD27 элиминировать солидную опухоль у мышей выявило более высокую противоопухолевую активность именно CAR-T-клеток с CD27 [52]. Наиболее эффективной оказалась комбинация сразу трех костимулирующих доменов – CD28, 4-1BB и CD27 – в составе CAR, что усилило пролиферацию, повысило устойчивость клеток к потере CAR, ослабило истощение CAR-T-клеток по сравнению с костимуляцией одним или двумя доменами [53, 54].
HVEM. Аббревиатура HVEM расшифровывается как Herpes Virus Entry Mediator, или медиатор проникновения вируса герпеса, так как первоначально эта молекула была открыта в качестве рецептора вируса простого герпеса-1 [55]. HVEM – это довольно необычный представитель своего суперсемейства, так как он связывает и молекулы, относящиеся к семейству TNFSF – TNFSF14 и лимфотоксин-α, и иммуноглобулин-подобные молекулы – CD272 и CD160 [56]. При этом HVEM костимулирует Т-клетки при транс-взаимодействиях, в то время как цис-взаимодействие препятствует костимуляции, формируя изолированный от других взаимодействий комплекс HVEM с CD272 или CD160 [57]. При активации HVEM связывает TRAF1, 2, 3, 5, что активирует передачу сигнала по путям NF-ϰB, JNK/AP-1 и PI3K/AKT и приводит к повышению синтеза как различных цитокинов, так и Bcl-2 [58, 59]. Это усиливает эффекторные свойства, пролиферацию и жизнеспособность Т-клеток. Молекулы TRAF, как предполагают, взаимодействуют с HVEM за счет мотива VTTVAVEET (рис. 3), который частично соответствует консервативному мотиву (P/A/S/T)X(E/Q)E [58].
Потенциал HVEM-зависимой костимуляции CAR-T-клеток оценили сравнительно недавно [60, 61]. Показано, что HVEM сочетает свойства рецепторов суперсемейств IgSF и TNFRSF. Так, если CD28 обеспечивает преимущественную дифференцировку модифицированных клеток в Т-клетки эффекторной памяти (Tem – effector memory T cell), а 4-1BB – в Tcm-клетки, то HVEM приводит к формированию сбалансированной популяции с практически одинаковыми долями как Tcm-, так и Tem-клеток. Кроме того, если костимуляция через CD28 активирует в основном гликолитический метаболизм, а через 4-1BB – окислительное фосфорилирование, то HVEM усиливает оба метаболических пути, формируя наиболее эффективное функциональное состояние CAR-T-клеток. Вовлечение в костимуляцию HVEM также способствует наименьшему истощению CAR-T-клеток по сравнению с CD28 и 4-1BB. CAR-T-клетки с HVEM показали наибольшую эффективность в случае солидных опухолей у мышей [61]. Кроме того, одновременная продукция CAR и лиганда HVEM TNFSF14 способствует проникновению CAR-T-клеток в опухоль за счет интенсивной секреции хемокинов [62].
TNFRSF18. TNFRSF18, больше известный как GITR (glucocorticoid-induced TNFR-related protein), конститутивно присутствует на низком уровне на мембране покоящихся Т-клеток. При активации уровень GITR на поверхности Т-клеток существенно возрастает. Уровень GITR в Тreg-клетках выше, чем в обычных Т-клетках, даже без стимуляции [63]. Взаимодействие GITR с лигандом GITRL ослабляет иммуносупрессивную активность Treg-клеток, а в эффекторных Т-клетках стимулирует пролиферацию и секрецию цитокинов, оказывает антиапоптотический эффект [64, 65]. Внутриклеточный сигналинг от GITR включает взаимодействие с TRAF1, 2, 3, 5 за счет мотивов STED и PEEE (рис. 3) [66]. Показано, что стимуляция T-клеток антителами к CD3, CD28 и GITR вызывает как сходные реакции, которые приводят к сигнальному синергизму в случае костимуляции, так и индивидуальные эффекты (например, стимуляция GITR вызывает усиленную продукцию ИЛ-27) [67]. Основные сигнальные пути, задействованные в костимуляции через GITR, – это NF-ϰB и MAPK [63].
Показано, что CAR-T-клетки, костимулированные через GITR, по эффективности уничтожения опухолей сравнимы с CAR-T-клетками на основе CD28 и 4-1BB [68, 69]. Кроме того, дополнительная продукция GITRL на CAR-T-клетках усиливает секрецию цитокинов, инфильтрацию опухолей и противоопухолевую активность [70].
Что касается других представителей TNFRSF, то появились работы по включению в состав CAR новых костимулирующих доменов рецепторов этого суперсемейства, например BAFF-R, CD30 и CD40 [35, 71, 72]. Костимуляция через CD40, как показано, ведет к более сильной активации пути NF-ϰB, чем костимуляция через 4-1BB, что может способствовать лучшей персистенции CD40 CAR-T-клеток in vivo.
Остальные костимулирующие домены
Костимулирующие молекулы, не входящие в суперсемейства иммуноглобулинов и рецепторов TNF, привлекают все большее внимание, так как интерес постепенно переключается на сигнальные процессы, происходящие в других иммунных клетках, таких как естественные киллеры, макрофаги и другие. Среди перспективных сигнальных молекул можно выделить Dap10 [73] и дектин-1 [74]. Современные технологии генной и клеточной инженерии позволяют достаточно легко создать библиотеки CAR с различными сочетаниями костимулирующих рецепторов или их частей [71, 75]. В сочетании с высокопроизводительным секвенированием это позволяет глубже оценить эффекты различных костимуляторов, не ограничиваясь наиболее изученными представителями семейств IgSF и TNFRSF, а также подобрать конкретные сочетания костимуляторов, ориентируясь на тип Т-клеток – CD4+ или CD8+.
СИГНАЛЬНЫЙ ДОМЕН CD3ζ И ЕГО АНАЛОГИ
На заре разработки в качестве сигнального домена в структуре CAR использовали только внутриклеточную часть CD3ζ [76, 77]. Это связано с концепцией самого рецептора, которая основывалась на комбинации B- и T-клеточных рецепторов для таргетного распознавания антигенов и последующей активации Т-клетки. Так, ранние исследования показали, что ICD CD3ζ подходит для активации Т-клеток, что заложило основу для конструирования CAR [78]. CD3ζ прочно «укоренился» в структуре рецептора и «кочевал» из поколения в поколение, обеспечивая основной сигнал активации для CAR-T-клеток [79]. Все одобренные к клиническому применению CAR-T-клеточные препараты несут в составе CAR именно CD3ζ (рис. 2), что подчеркивает важность этого домена для разработчиков и, как казалось до недавнего времени, отсутствие альтернатив [5].
Остальные белки группы CD3
Однако интерес к этой части CAR значительно повысился со временем. Так, в 2018 году Sadelain и соавт. показали, что для полноценного функционирования CAR в CD3ζ достаточно оставить один активный иммунорецепторный тирозиновый активирующий мотив (ITAM – immunoreceptor tyrosine-based activation motif) из трех [80]. Важную роль при этом играет как его расположение, так и аминокислотный состав. Наиболее функциональным вариантом для элиминирования опухолей оказался 1XX (1 – позиция активного ITAM относительно мембраны клетки, X – неактивный ITAM), в то время как XX3 умеренно поддерживал персистенцию CAR-T-клеток. Эти данные подчеркнули, что необходимо пересмотреть отношение к казалось бы незаменимому CD3ζ.
В результате были проведены исследования возможных аналогов CD3ζ, а именно, других представителей группы CD3 – ε, δ, γ [81, 82]. В отличие от CD3ζ, эти молекулы в ICD содержат только один ITAM [83]. Хотя все ITAM содержат консервативную последовательность YXXL/I-X6–8-YXXL/I (X – любая АК), состав АК каждого ITAM уникален, что определяет различия в аффинности связывания сигнальных молекул (рис. 4) [84].

Рис. 4. Структура и функциональные особенности белков группы CD3. Показана организация внутриклеточных доменов CD3ε, δ, ζ, γ, которые содержат уникальные мотивы для взаимодействия с внутриклеточными сигнальными партнерами. ζ, γ, δ, ε – представители группы CD3 Т-клеточного рецептора; ITAM – иммунорецепторный тирозиновый активирующий мотив; BRS – участок, богатый основными аминокислотами; PRS – участок, богатый пролином; RK – мотив рецепторной киназы; LL – серинзависимый дилейциновый мотив (* указывает на отсутствие серина перед LL, что снижает вовлечение LL в регуляцию Т-клеточного рецептора); ТКР – Т-клеточный рецептор; α, β – распознающие цепи Т-клеточного рецептора; КМ – клеточная мембрана; Y – тирозин; pY – фосфорилированный тирозин
Всего мультисубъединичный комплекс ТКР–CD3 содержит 10 ITAM. Высокая концентрация тирозиновых мотивов, вероятно, способствует усилению сигнала, поскольку уменьшение их числа приводит к нарушению функции комплекса ТКР–CD3 у мышей [85]. Кроме того, различия между CD3 и ITAM, содержащихся в них, также важно для передачи сигнала и развития зрелых Т-клеток [86].
Помимо уникальных аминокислотных последовательностей в ITAM, внутриклеточные части каждой субъединицы CD3 имеют свои особенности (рис. 4). CD3ζ и CD3ε содержат участки, обогащенные положительно заряженными АК (BRS – basic-rich stretch), с помощью которых взаимодействуют с внутренней стороной клеточной мембраны [87, 88]. CD3ε взаимодействует с киназой LCK либо через ионные связи между BRS и кислыми остатками в уникальном домене LCK, либо с помощью мотива рецепторной киназы (RK – receptor kinase) и SH3-домена LCK [89, 90]. CD3ε также содержит богатую пролином последовательность (PRS – proline-rich sequence), которая взаимодействует с адаптерным белком NCK, что необходимо для созревания ИС и активации Т-клеток [91]. В CD3γ присутствует проксимальный серинзависимый дилейциновый (SDKQTLL) мотив, который участвует в уменьшении количества ТКР на мембране клетки посредством механизма, зависимого от протеинкиназы C (PKC – protein kinase C) [92]. В CD3δ, кроме ITAM, также есть похожий мотив (ADTQALL), в котором, однако, отсутствует серин, требующийся для взаимодействия с PKC, поэтому CD3δ считается менее значимым для регуляции количества ТКР на мембране в отличие от CD3γ [93].
Уникальные мотивы в структуре каждого представителя белков CD3 играют важную роль в контексте CAR, хотя каждый вариант CD3 по отдельности может быть достаточным для создания функциональной структуры CAR. Это показано путем включения CD3ε, δ или γ в структуру CAR в качестве сигнального домена вместо CD3ζ [81, 82]. Обнаружено, что CD3δ, CD3ε или CD3γ CAR-T-клетки более эффективно элиминируют опухоли in vivo по сравнению с CD3ζ. Это связано с уникальными особенностями конкретных представителей группы CD3. Так, ICD CD3ε связывает киназу CSK, которая ингибирует активацию киназы LCK, что снижает истощение CAR-T-клеток и способствует их персистенции; монофосфорилированный по второму тирозину ICD CD3δ связывает фосфатазу SHP-1, что также снижает интенсивность сигналинга и секрецию цитокинов, вероятно, за счет ослабления активации пути NF-ϰB. Результаты транскриптомного анализа свидетельствуют о снижении экспрессии генов, продукты которых участвуют в гликолизе, повышении экспрессии генов, продукты которых вовлечены в митохондриальный метаболизм, что характеризует фенотип Tm-клеток. Кроме того, в случае CD3δ интенсивно экспрессируется TCF-1, ассоциированный со стволовыми T-клетками памяти [94]. Клетки этого типа, наряду с Tn-клетками, обладают наибольшим потенциалом к самообновлению и способны дифференцироваться в любой тип клеток памяти [95]. Также, вероятно, CD3ε, который обладает большим соотношением положительно заряженных и кислых АК, связывается с мембранными фосфолипидами с более высокой интенсивностью, чем CD3ζ, снижая доступность CAR для внутриклеточных сигнальных партнеров, что наблюдается в случае ТКР и других белков [88, 96]. Это, в свою очередь, снижает вероятность возникновения неспецифического и тонического сигналинга. За счет использования в структуре CAR шарнирного домена из CD8α, способного к димеризации, удалось также показать, что димерные формы CD3δ и CD3γ усиливают секрецию цитокинов CAR-T-клетками, а также количество поверхностных CD69 и 4-1BB, особенно в случае мутации дилейциновых мотивов (SDKQTAL и ADTQAAL) [81].
Сигнальные партнеры ТКР
Новый формат CAR, названных bypassCAR (bCAR), появился благодаря изучению отдельных элементов сигнальных каскадов при активации ТКР. В структуру bCAR были интегрированы части внутриклеточных сигнальных партнеров ТКР вместо доменов, содержащих ITAM. Первые bCAR-подобные химерные молекулы были созданы в конце прошлого века для определения ключевых киназ, необходимых для активации T-клеток [97]. В структуре таких рецепторов CD16 использовали в сочетании с LCK, FYN, SYK или ZAP70. При этом только в случае домена SYK модифицированные клетки могли лизировать клетки-мишени в ответ на стимуляцию. При замене CD16 на scFv к соответствующему антигену сохранялась уникальная способность SYK активировать bCAR-T-клетку, минуя ТКР [98].
Позднее была сконструирована панель противоопухолевых bCAR с CSK, FYN, киназным доменом ZAP70 (ZAP70KD – ZAP70 kinase domain), LAT, SLP76 или PLCγ1 без костимулирующих доменов [99]. bCAR на основе ZAP70KD и PLCγ1 активировали модифицированные T-клетки, хотя PLCγ1 bCAR экспрессировался значительно слабее. В in vivo экспериментах ZAP70KD bCAR-T-клетки эффективнее элиминировали солидную опухоль, чем CD3ζ CAR-T-клетки с костимулирующим доменом 4-1BB. bCAR на основе ZAP70KD активировал T-клетки с нокаутом ТКР и LCK, но не в отсутствие SLP76 или LAT, что подтверждает сохранение структуры сигнальных путей после ТКР.
Недавно были разработаны bCAR второго поколения, где дополнительно встроили адаптерный домен из LAT или SLP76 по аналогии с костимулирующими доменами традиционных CAR. Однако Т-клетки, модифицированные такими конструкциями, обладали чрезмерно высоким уровнем тонического сигналинга [100]. При этом добавление сигнального домена CD28 перед киназным доменом ZAP70 привело к более длительной ремиссии B-клеточной опухоли у мышей под действием bCAR-T-клеток по сравнению с традиционными CAR-T-клетками второго поколения с CD3ζ и костимулирующим доменом CD28.
ЗАКЛЮЧЕНИЕ
Исследование комбинаций костимулирующих и сигнальных доменов CAR – это динамично развивающееся направление изучения механизмов функционирования CAR-T-клеток и расширения возможностей применения этих клеток. Разнообразие этих доменов открывает широкие возможности для конструирования CAR-T-клеток нового поколения с улучшенными функциональными характеристиками.
Анализ накопленных данных демонстрирует, что выбор того или иного костимулирующего домена оказывает значительное влияние не только на активацию и цитотоксическую активность CAR-T-клеток, но и определяет тип их метаболической активности, способность к долговременной персистенции in vivo и устойчивость к функциональному истощению. При этом комбинация различных доменов или создание модульных конструкций может позволить преодолеть ключевые ограничения текущих подходов к созданию терапевтических CAR-T-продуктов и их применения, таких как гетерогенность опухолевых антигенов, иммуносупрессивное микроокружение и ассоциированную с адоптивным переносом токсичность.
Кроме того, существует потребность в уменьшении размера CAR с сохранением функциональности и поиске минимально активной структуры рецептора, что должно благоприятствовать успеху модификации и способствовать повышенной и стабильной продукции рецептора CAR-Т-клетками. Такие исследования тоже ведутся [101].
Дальнейшая оптимизация сигнальных доменов CAR-T-клеток требует не только углубленного понимания молекулярных механизмов активации Т-лимфоцитов, но и применения передовых технологий, включая CRISPR-скрининг, транскриптомику, протеомику и компьютерное моделирование [75, 102–105]. Это позволит создавать персонализированные клеточные продукты, максимально адаптированные к биологии конкретного типа опухоли. Наряду с совершенствованием поколений рецептора и созданием модульных систем [106–108], исследования в этой области могут привести к прорывным терапевтическим решениям, расширяющим спектр применения технологии CAR-T и повышающим ее эффективность при лечении как онкологических, так и, возможно, аутоиммунных и инфекционных заболеваний.
Работа выполнена при поддержке Министерства науки и высшего образования РФ (соглашение № 075-15-2024-536).
Об авторах
Д. В. Волков
ГНЦ Институт биоорганической химии им. академиков М.М. Шемякина и Ю.А. Овчинникова РАН
Автор, ответственный за переписку.
Email: ya.wolf.otl@yandex.ru
Россия, Москва, 117997
В. М. Степанова
ГНЦ Институт биоорганической химии им. академиков М.М. Шемякина и Ю.А. Овчинникова РАН
Email: ya.wolf.otl@yandex.ru
Россия, Москва, 117997
И. А. Ярошевич
ГНЦ Институт биоорганической химии им. академиков М.М. Шемякина и Ю.А. Овчинникова РАН; Московский государственный университет имени М.В. Ломоносова
Email: ya.wolf.otl@yandex.ru
Россия, Москва, 117997; Москва 119991
А. Г. Габибов
ГНЦ Институт биоорганической химии им. академиков М.М. Шемякина и Ю.А. Овчинникова РАН
Email: ya.wolf.otl@yandex.ru
Россия, Москва, 117997
Ю. П. Рубцов
ГНЦ Институт биоорганической химии им. академиков М.М. Шемякина и Ю.А. Овчинникова РАН
Email: ya.wolf.otl@yandex.ru
Россия, Москва, 117997
Список литературы
- Yazbeck V, Alesi E, Myers J, et al. An overview of chemotoxicity and radiation toxicity in cancer therapy. Adv. Cancer Res. 2022:1–27.
- Cappell KM, Kochenderfer JN. Long-term outcomes following CAR T cell therapy: what we know so far. Nat Rev Clin Oncol. 2023;20(6):359–371. doi: 10.1038/s41571-023-00754-1
- Li Q, Lei X, Zhu J, et al. Radiotherapy/chemotherapy-immunotherapy for cancer management: From mechanisms to clinical implications. Oxid Med Cell Longev. 2023;2023:7530794. doi: 10.1155/2023/7530794
- McLellan AD, Ali Hosseini Rad SM. Chimeric antigen receptor T cell persistence and memory cell formation. Immunol Cell Biol. 2019;97(7):664–674. doi: 10.1111/imcb.12254
- Mitra A, Barua A, Huang L, et al. From bench to bedside: the history and progress of CAR T cell therapy. Front Immunol. 2023;14:1188049. doi: 10.3389/fimmu.2023.1188049
- Finney HM, Lawson ADG, Bebbington CR, Weir ANC. Chimeric receptors providing both primary and costimulatory signaling in T cells from a single gene product. J Immunol. 1998;161(6):2791–2797. doi: 10.4049/jimmunol.161.6.2791
- Krause A, Guo HF, Latouche JB, et al. Antigen-dependent CD28 signaling selectively enhances survival and proliferation in genetically modified activated human primary T lymphocytes. J Exp Med. 1998;188(4):619–626. doi: 10.1084/jem.188.4.619
- Esensten JH, Helou YA, Chopra G, et al. CD28 costimulation: From mechanism to therapy. Immunity. 2016;44(5):973–988. doi: 10.1016/j.immuni.2016.04.020
- Honikel MM, Olejniczak SH. Co-stimulatory receptor signaling in CAR-T cells. Biomolecules. 2022;12(9):1303. doi: 10.3390/biom12091303
- Kunkl M, Sambucci M, Ruggieri S, et al. CD28 autonomous signaling up-regulates C-myc expression and promotes glycolysis enabling inflammatory T cell responses in multiple sclerosis. Cells. 2019;8(6):575. doi: 10.3390/cells8060575
- Kawalekar OU, O’Connor RS, Fraietta JA, et al. Distinct signaling of coreceptors regulates specific metabolism pathways and impacts memory development in CAR T cells. Immunity. 2016;44(2):380–390. doi: 10.1016/j.immuni.2016.01.021
- Guedan S, Madar A, Casado-Medrano V, et al. Single residue in CD28-costimulated CAR-T cells limits long-term persistence and antitumor durability. J Clin Invest. 2020;130(6):3087–3097. doi: 10.1172/JCI133215
- Boucher JC, Li G, Kotani H, et al. CD28 costimulatory domain-targeted mutations enhance chimeric antigen receptor T-cell function. Cancer Immunol Res. 2021;9(1):62–74. doi: 10.1158/2326-6066.CIR-20-0253
- Kofler DM, Chmielewski M, Rappl G, et al. CD28 costimulation Impairs the efficacy of a redirected t-cell antitumor attack in the presence of regulatory t cells which can be overcome by preventing Lck activation. Mol Ther. 2011;19(4):760–767. doi: 10.1038/mt.2011.9
- Gulati P, Rühl J, Kannan A, et al. Aberrant lck signal via CD28 costimulation augments antigen-specific functionality and tumor control by redirected T cells with PD-1 blockade in humanized mice. Clin Cancer Res. 2018;24(16):3981-3993. doi: 10.1158/1078-0432.ccr-17-1788
- Ferreira LMR, Muller YD. CAR T-cell therapy: Is CD28-CAR heterodimerization its Achilles’ heel? Front Immunol. 2021;12:766220. doi: 10.3389/fimmu.2021.766220
- Yoshinaga SK, Whoriskey JS, Khare SD, et al. T-cell co-stimulation through B7RP-1 and ICOS. Nature. 1999;402(6763):827–832. doi: 10.1038/45582
- van Berkel MEAT, Oosterwegel MA. CD28 and ICOS: similar or separate costimulators of T cells? Immunol Lett. 2006;105(2):115–122. doi: 10.1016/j.imlet.2006.02.007
- Paulos CM, Carpenito C, Plesa G, et al. The inducible costimulator (ICOS) is critical for the development of human TH17 cells. Sci Transl Med. 2010;2(55):55ra78-55ra78. doi: 10.1126/scitranslmed.3000448
- Parry RV, Rumbley CA, Vandenberghe LH, et al. CD28 and inducible costimulatory protein Src homology 2 binding domains show distinct regulation of phosphatidylinositol 3-kinase, Bcl-xL, and IL-2 expression in primary human CD4 T lymphocytes. J Immunol. 2003;171(1):166–174. doi: 10.4049/jimmunol.171.1.166
- Fos C, Salles A, Lang V, et al. ICOS ligation recruits the p50alpha PI3K regulatory subunit to the immunological synapse. J Immunol. 2008;181(3):1969-1977. doi: 10.4049/jimmunol.181.3.1969
- Shen C-J, Yang Y-X, Han EQ, et al. Chimeric antigen receptor containing ICOS signaling domain mediates specific and efficient antitumor effect of T cells against EGFRvIII expressing glioma. J Hematol Oncol. 2013;6(1):33. doi: 10.1186/1756-8722-6-33
- Guedan S, Chen X, Madar A, et al. ICOS-based chimeric antigen receptors program bipolar TH17/TH1 cells. Blood. 2014;124(7):1070–1080. doi: 10.1182/blood-2013-10-535245
- Wyatt MM, Huff LW, Nelson MH, et al. Augmenting TCR signal strength and ICOS costimulation results in metabolically fit and therapeutically potent human CAR Th17 cells. Mol Ther. 2023;31(7):2120–2131. doi: 10.1016/j.ymthe.2023.04.010
- Fujiwara K, Kitaura M, Tsunei A, et al. Structure of the signal transduction domain in second-generation CAR regulates the input efficiency of CAR signals. Int J Mol Sci. 2021;22(5):2476. doi: 10.3390/ijms22052476
- Vanamee ÉS, Faustman DL. Structural principles of tumor necrosis factor superfamily signaling. Sci Signal. 2018;11(511):eaao4910. doi: 10.1126/scisignal.aao4910
- Ye H, Park YC, Kreishman M, et al. The structural basis for the recognition of diverse receptor sequences by TRAF2. Mol Cell. 1999;4(3):321–330. doi: 10.1016/s1097-2765(00)80334-2
- Ward-Kavanagh LK, Lin WW, Šedý JR, Ware CF. The TNF receptor superfamily in co-stimulating and co-inhibitory responses. Immunity. 2016;44(5):1005–1019. doi: 10.1016/j.immuni.2016.04.019
- Craxton A, Draves KE, Gruppi A, Clark EA. BAFF regulates B cell survival by downregulating the BH3-only family member Bim via the ERK pathway. J Exp Med. 2005;202(10):1363–1374. doi: 10.1084/jem.20051283
- Teijeira A, Labiano S, Garasa S, et al. Mitochondrial morphological and functional reprogramming following CD137 (4-1BB) costimulation. Cancer Immunol Res. 2018;6(7):798–811. doi: 10.1158/2326-6066.cir-17-0767
- Glez-Vaz J, Azpilikueta A, Ochoa MC, et al. CD137 (4-1BB) requires physically associated cIAPs for signal transduction and antitumor effects. Sci Adv. 2023;9(33):eadf6692. doi: 10.1126/sciadv.adf6692
- Cappell KM, Kochenderfer JN. A comparison of chimeric antigen receptors containing CD28 versus 4-1BB costimulatory domains. Nat Rev Clin Oncol. 2021;18(11):715–727. doi: 10.1038/s41571-021-00530-z
- Boroughs AC, Larson RC, Marjanovic ND, et al. A distinct transcriptional program in human CAR T cells bearing the 4-1BB signaling domain revealed by scRNA-seq. Mol Ther. 2020;28(12):2577–2592. doi: 10.1016/j.ymthe.2020.07.023
- Sun C, Shou P, Du H, et al. THEMIS-SHP1 recruitment by 4-1BB tunes LCK-mediated priming of chimeric antigen receptor-redirected T cells. Cancer Cell. 2020;37(2):216–225.e6. doi: 10.1016/j.ccell.2019.12.014
- Mamonkin M, Mukherjee M, Srinivasan M, et al. Reversible transgene expression reduces fratricide and permits 4-1BB costimulation of CAR T cells directed to T-cell malignancies. Cancer Immunol Res. 2018;6(1):47–58. doi: 10.1158/2326-6066.CIR-17-0126
- Dou Z, Bonacci TR, Shou P, et al. 4-1BB-encoding CAR causes cell death via sequestration of the ubiquitin-modifying enzyme A20. Cell Mol Immunol. 2024;21(8):905–917. doi: 10.1038/s41423-024-01198-y
- Gomes-Silva D, Mukherjee M, Srinivasan M, et al. Tonic 4-1BB costimulation in chimeric antigen receptors impedes T cell survival and is vector-dependent. Cell Rep. 2017;21(1):17–26. doi: 10.1016/j.celrep.2017.09.015
- Willoughby J, Griffiths J, Tews I, Cragg MS. OX40: Structure and function - What questions remain? Mol Immunol. 2017;83:13–22. doi: 10.1016/j.molimm.2017.01.006
- Croft M. Costimulation of T cells by OX40, 4-1BB, and CD27. Cytokine Growth Factor Rev. 2003;14(3-4):265–273. doi: 10.1016/s1359-6101(03)00025-x
- Kawamata S, Hori T, Imura A, et al. Activation of OX40 signal transduction pathways leads to tumor necrosis factor receptor-associated factor (TRAF) 2- and TRAF5-mediated NF-κB activation. J Biol Chem. 1998;273(10):5808–5814. doi: 10.1074/jbc.273.10.5808
- Croft M. Control of immunity by the TNFR-related molecule OX40 (CD134). Annu Rev Immunol. 2010;28(1):57–78. doi: 10.1146/annurev-immunol-030409-101243
- Song J, So T, Croft M. Activation of NF-kappaB1 by OX40 contributes to antigen-driven T cell expansion and survival. J Immunol. 2008;180(11):7240–7248. doi: 10.4049/jimmunol.180.11.7240
- Tan J, Jia Y, Zhou M, et al. Chimeric antigen receptors containing the OX40 signalling domain enhance the persistence of T cells even under repeated stimulation with multiple myeloma target cells. J Hematol Oncol. 2022;15(1):39. doi: 10.1186/s13045-022-01244-0
- Starzer AM, Berghoff AS. New emerging targets in cancer immunotherapy: CD27 (TNFRSF7). ESMO Open. 2020;4(Suppl 3):e000629. doi: 10.1136/esmoopen-2019-000629
- Akiba H, Nakano H, Nishinaka S, et al. CD27, a member of the tumor necrosis factor receptor superfamily, activates NF-kappaB and stress-activated protein kinase/c-Jun N-terminal kinase via TRAF2, TRAF5, and NF-kappaB-inducing kinase. J Biol Chem. 1998;273(21):13353–13358. doi: 10.1074/jbc.273.21.13353
- Dolfi DV, Boesteanu AC, Petrovas C, et al. Late signals from CD27 prevent Fas-dependent apoptosis of primary CD8+ T cells. J Immunol. 2008;180(5):2912–2921. doi: 10.4049/jimmunol.180.5.2912
- van de Ven K, Borst J. Targeting the T-cell co-stimulatory CD27/CD70 pathway in cancer immunotherapy: rationale and potential. Immunotherapy. 2015;7(6):655–667. doi: 10.2217/imt.15.32
- Peperzak V, Veraar EAM, Keller AM, et al. The Pim kinase pathway contributes to survival signaling in primed CD8+ T cells upon CD27 costimulation. J Immunol. 2010;185(11):6670–6678. doi: 10.4049/jimmunol.1000159
- Yamamoto H, Kishimoto T, Minamoto S. NF-κB activation in CD27 signaling: Involvement of TNF receptor-associated factors in its signaling and identification of functional region of CD27. J Immunol. 1998;161(9):4753–4759. doi: 10.4049/jimmunol.161.9.4753
- Song D-G, Ye Q, Poussin M, et al. CD27 costimulation augments the survival and antitumor activity of redirected human T cells in vivo. Blood. 2012;119(3):696–706. doi: 10.1182/blood-2011-03-344275
- Song D-G, Powell DJ. Pro-survival signaling via CD27 costimulation drives effective CAR T-cell therapy. Oncoimmunology. 2012;1(4):547–549. doi: 10.4161/onci.19458
- Han Y, Xie W, Song D-G, Powell DJ, Jr. Control of triple-negative breast cancer using ex vivo self-enriched, costimulated NKG2D CAR T cells. J Hematol Oncol. 2018;11(1):92. doi: 10.1186/s13045-018-0635-z
- Zhang C, Jia J, Heng G, et al. CD27 agonism coordinates with CD28 and 4-1BB signal to augment the efficacy of CAR-T cells in colorectal tumor. Med Oncol. 2023;40(4):123. doi: 10.1007/s12032-023-01959-1
- Supimon K, Sangsuwannukul T, Sujjitjoon J, et al. Anti-mucin 1 chimeric antigen receptor T cells for adoptive T cell therapy of cholangiocarcinoma. Sci Rep. 2021;11(1):6276. doi: 10.1038/s41598-021-85747-9
- Montgomery RI, Warner MS, Lum BJ, Spear PG. Herpes simplex virus-1 entry into cells mediated by a novel member of the TNF/NGF receptor family. Cell. 1996;87(3):427–436. doi: 10.1016/s0092-8674(00)81363-x
- Šedý JR, Ramezani-Rad P. HVEM network signaling in cancer. Advances in Cancer Research. 2019;142:145–186. doi: 10.1016/bs.acr.2019.01.004
- Steinberg MW, Cheung TC, Ware CF. The signaling networks of the herpesvirus entry mediator (TNFRSF14) in immune regulation. Immunol Rev. 2011;244(1):169–187. doi: 10.1111/j.1600-065X.2011.01064.x
- Hsu H, Solovyev I, Colombero A, et al. ATAR, a novel tumor necrosis factor receptor family member, signals through TRAF2 and TRAF5. J Biol Chem. 1997;272(21):13471–13474. doi: 10.1074/jbc.272.21.13471
- Soroosh P, Doherty TA, So T, et al. Herpesvirus entry mediator (TNFRSF14) regulates the persistence of T helper memory cell populations. J Exp Med. 2011;208(4):797–809. doi: 10.1084/jem.20101562
- Nunoya J-I, Masuda M, Ye C, Su L. Chimeric antigen receptor T cell bearing herpes virus entry mediator co-stimulatory signal domain exhibits high functional potency. Mol Ther Oncolytics. 2019;14:27–37. doi: 10.1016/j.omto.2019.03.002
- Sun S, Huang C, Lu M, et al. Herpes virus entry mediator costimulation signaling enhances CAR T-cell efficacy against solid tumors through metabolic reprogramming. Cancer Immunol Res. 2023;11(4):515–529. doi: 10.1158/2326-6066.CIR-22-0531
- Zhang N, Liu X, Qin J, et al. LIGHT/TNFSF14 promotes CAR-T cell trafficking and cytotoxicity through reversing immunosuppressive tumor microenvironment. Mol Ther. 2023;31(9):2575–2590. doi: 10.1016/j.ymthe.2023.06.015
- Azuma M. Co-signal molecules in T-cell activation: Historical Overview and Perspective. Adv Exp Med Biol. 2019;1189:3–23. doi: 10.1007/978-981-32-9717-3_1
- Tian J, Zhang B, Rui K, Wang S. The role of GITR/GITRL interaction in autoimmune diseases. Front Immunol. 2020;11:588682. doi: 10.3389/fimmu.2020.588682
- Ronchetti S, Zollo O, Bruscoli S, et al. GITR, a member of the TNF receptor superfamily, is costimulatory to mouse T lymphocyte subpopulations. Eur J Immunol. 2004;34(3):613–622. doi: 10.1002/eji.200324804
- So T, Nagashima H, Ishii N. TNF receptor-associated factor (TRAF) signaling network in CD4(+) T-lymphocytes. Tohoku J Exp Med. 2015;236(2):139–154. doi: 10.1620/tjem.236.139
- Kanamaru F, Youngnak P, Hashiguchi M, et al. Costimulation via glucocorticoid-induced TNF receptor in both conventional and CD25+ regulatory CD4+ T cells. J Immunol. 2004;172(12):7306–7314. doi: 10.4049/jimmunol.172.12.7306
- Xi B, Berahovich R, Zhou H, et al. A real-time potency assay for chimeric antigen receptor T cells targeting solid and hematological cancer cells. J Vis Exp. 2019;153. doi: 10.3791/59033
- Golubovskaya VM. GITR domain inside CAR co-stimulates activity of CAR-T cells against cancer. Front Biosci. 2018;23(12):2245–2254. doi: 10.2741/4703
- Wang Y, Wang L, Seo N, et al. CAR-modified Vγ9Vδ2 T cells propagated using a novel bisphosphonate prodrug for allogeneic adoptive immunotherapy. Int J Mol Sci. 2023;24(13):10873. doi: 10.3390/ijms241310873
- Goodman DB, Azimi CS, Kearns K, et al. Pooled screening of CAR T cells identifies diverse immune signaling domains for next-generation immunotherapies. Sci Transl Med. 2022;14(670):eabm1463. doi: 10.1126/scitranslmed.abm1463
- Levin-Piaeda O, Levin N, Pozner S, et al. The intracellular domain of CD40 is a potent costimulatory element in chimeric antigen receptors. J Immunother. 2021;44(6):209–213. doi: 10.1097/CJI.0000000000000373
- Li S, Zhao R, Zheng D, et al. DAP10 integration in CAR-T cells enhances the killing of heterogeneous tumors by harnessing endogenous NKG2D. Mol Ther Oncolytics. 2022;26:15–26. doi: 10.1016/j.omto.2022.06.003
- Liang X, Huang Y, Li D, et al. Distinct functions of CAR-T cells possessing a dectin-1 intracellular signaling domain. Gene Ther. 2023/5 2023;30(5):411–420. doi: 10.1038/s41434-021-00257-7
- Daniels KG, Wang S, Simic MS, et al. Decoding CAR T cell phenotype using combinatorial signaling motif libraries and machine learning. Science. 2022;378(6625):1194–1200. doi: 10.1126/science.abq0225
- Gross G, Waks T, Eshhar Z. Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc Natl Acad Sci U S A. 1989;86(24):10024–10028. doi: 10.1073/pnas.86.24.10024
- Kuwana Y, Asakura Y, Utsunomiya N, et al. Expression of chimeric receptor composed of immunoglobulin-derived V resions and T-cell receptor-derived C regions. Biochem Biophys Res Commun. 1987;149(3):960–968. doi: 10.1016/0006-291x(87)90502-x
- Eshhar Z, Waks T, Gross G, Schindler DG. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta subunits of the immunoglobulin and T-cell receptors. Proc Natl Acad Sci U S A. 1993;90(2):720–724. doi: 10.1073/pnas.90.2.720
- Zheng Z, Li S, Liu M, et al. Fine-tuning through generations: Advances in structure and production of CAR-T therapy. Cancers (Basel). 2023;15(13):3476. doi: 10.3390/cancers15133476
- Feucht J, Sun J, Eyquem J, et al. Calibrated CAR activation potential directs alternative T cell fates and therapeutic potency. Blood. 2018;132(Supplement 1):1412–1412. doi: 10.1182/blood-2018-99-117698
- Velasco Cárdenas RMH, Brandl SM, Meléndez AV, et al. Harnessing CD3 diversity to optimize CAR T cells. Nat Immunol. 2023;24(12):2135–2149. doi: 10.1038/s41590-023-01658-z
- Wang P, Wang Y, Zhao X, et al. Chimeric antigen receptor with novel intracellular modules improves antitumor performance of T cells. Signal Transduct Target Ther. 2025;10(1):20. doi: 10.1038/s41392-024-02096-5
- Reth M. Antigen receptor tail clue. Nature. 1989;338(6214):383–384. doi: 10.1038/338383b0
- Love PE, Hayes SM. ITAM-mediated signaling by the T-cell antigen receptor. Cold Spring Harb Perspect Biol. 2010;2(6):a002485. doi: 10.1101/cshperspect.a002485
- Holst J, Wang H, Eder KD, et al. Scalable signaling mediated by T cell antigen receptor-CD3 ITAMs ensures effective negative selection and prevents autoimmunity. Nat Immunol. 2008;9(6):658–666. doi: 10.1038/ni.1611
- Bettini ML, Chou P-C, Guy CS, et al. Cutting edge: CD3 ITAM diversity is required for optimal TCR signaling and thymocyte development. J Immunol. 2017;199(5):1555–1560. doi: 10.4049/jimmunol.1700069
- Aivazian D, Stern LJ. Phosphorylation of T cell receptor zeta is regulated by a lipid dependent folding transition. Nat Struct Biol. 2000;7(11):1023–1026. doi: 10.1038/80930
- Xu C, Gagnon E, Call ME, et al. Regulation of T cell receptor activation by dynamic membrane binding of the CD3ε cytoplasmic tyrosine-based motif. Cell. 2008;135(4):702–713. doi: 10.1016/j.cell.2008.09.044
- Li L, Guo X, Shi X, et al. Ionic CD3−Lck interaction regulates the initiation of T-cell receptor signaling. Proc Natl Acad Sci U S A. 2017;114(29):E5891–E5899. doi: 10.1073/pnas.1701990114
- Hartl FA, Beck-Garcìa E, Woessner NM, et al. Noncanonical binding of Lck to CD3ε promotes TCR signaling and CAR function. Nat Immunol. 2020;21(8):902–913. doi: 10.1038/s41590-020-0732-3
- Gil D, Schamel WWA, Montoya Ma, et al. Recruitment of nck by CD3ε reveals a ligand-induced conformational change essential for T cell receptor signaling and synapse formation. Cell. 2002;109(7):901–912. doi: 10.1016/s0092-8674(02)00799-7
- Dietrich J, Hou X, Wegener AM, Geisler C. CD3 gamma contains a phosphoserine-dependent di-leucine motif involved in down-regulation of the T cell receptor. EMBO J. 1994;13(9):2156–2166. doi: 10.1002/j.1460-2075.1994.tb06492.x
- Wegener A-MK, Hou X, Dietrich J, Geisler C. Distinct domains of the CD3-γ chain are involved in surface expression and function of the T cell antigen receptor. J Biol Chem. 1995;270(9):4675–4680. doi: 10.1074/jbc.270.9.4675
- Escobar G, Mangani D, Anderson AC. T cell factor 1: A master regulator of the T cell response in disease. Sci Immunol. 2020;5(53):eabb9726. doi: 10.1126/sciimmunol.abb9726
- Gattinoni L, Speiser DE, Lichterfeld M, Bonini C. T memory stem cells in health and disease. Nat Med. 2017;23(1):18–27. doi: 10.1038/nm.4241
- Yeung T, Gilbert GE, Shi J, et al. Membrane phosphatidylserine regulates surface charge and protein localization. Science. 2008;319(5860):210–213. doi: 10.1126/science.1152066
- Kolanus W, Romeo C, Seed B. T cell activation by clustered tyrosine kinases. Cell. 1993;74(1):171–183. doi: 10.1016/0092-8674(93)90304-9
- Fitzer-Attas CJ, Schindler DG, Waks T, Eshhar Z. Harnessing Syk family tyrosine kinases as signaling domains for chimeric single chain of the variable domain receptors: optimal design for T cell activation. J Immunol. 1998;160(1):145–154. doi: 10.4049/jimmunol.160.1.145
- Tousley AM, Rotiroti MC, Labanieh L, et al. Co-opting signalling molecules enables logic-gated control of CAR T cells. Nature. 2023;615(7952):507–516. doi: 10.1038/s41586-023-05778-2
- Balagopalan L, Moreno T, Qin H, et al. Generation of antitumor chimeric antigen receptors incorporating T cell signaling motifs. Sci Signal. 2024;17(846):eadp8569. doi: 10.1126/scisignal.adp8569
- Si W, Fan Y-Y, Qiu S-Z, et al. Design of diversified chimeric antigen receptors through rational module recombination. iScience. 2023;26(4):106529. doi: 10.1016/j.isci.2023.106529
- Salter AI, Ivey RG, Kennedy JJ, et al. Phosphoproteomic analysis of chimeric antigen receptor signaling reveals kinetic and quantitative differences that affect cell function. Sci Signal. 2018;11(544):eaat6753. doi: 10.1126/scisignal.aat6753
- Ramello MC, Benzaïd I, Kuenzi BM, et al. An immunoproteomic approach to characterize the CAR interactome and signalosome. Sci Signal. 2019;12(568):eaap9777. doi: 10.1126/scisignal.aap9777
- Qiu S, Chen J, Wu T, et al. CAR-Toner: an AI-driven approach for CAR tonic signaling prediction and optimization. Cell Res. 2024;34(5):386–388. doi: 10.1038/s41422-024-00936-1
- Rohrs JA, Zheng D, Graham NA, et al. Computational model of chimeric antigen receptors explains site-specific phosphorylation kinetics. Biophys J. 2018;115(6):1116–1129. doi: 10.1016/j.bpj.2018.08.018
- Sheykhhasan M, Ahmadieh-Yazdi A, Vicidomini R, et al. CAR T therapies in multiple myeloma: unleashing the future. Cancer Gene Ther. 2024;31(5):667–686. doi: 10.1038/s41417-024-00750-2
- Stepanov AV, Xie J, Zhu Q, et al. Control of the antitumour activity and specificity of CAR T cells via organic adapters covalently tethering the CAR to tumour cells. Nat Biomed Eng. 2024;8(5):529–543. doi: 10.1038/s41551-023-01102-5
- Stepanov AV, Kalinin RS, Shipunova VO, et al. Switchable targeting of solid tumors by BsCAR T cells. Proc Natl Acad Sci U S A. 2022;119(46):e2210562119. doi: 10.1073/pnas.2210562119
Дополнительные файлы

